
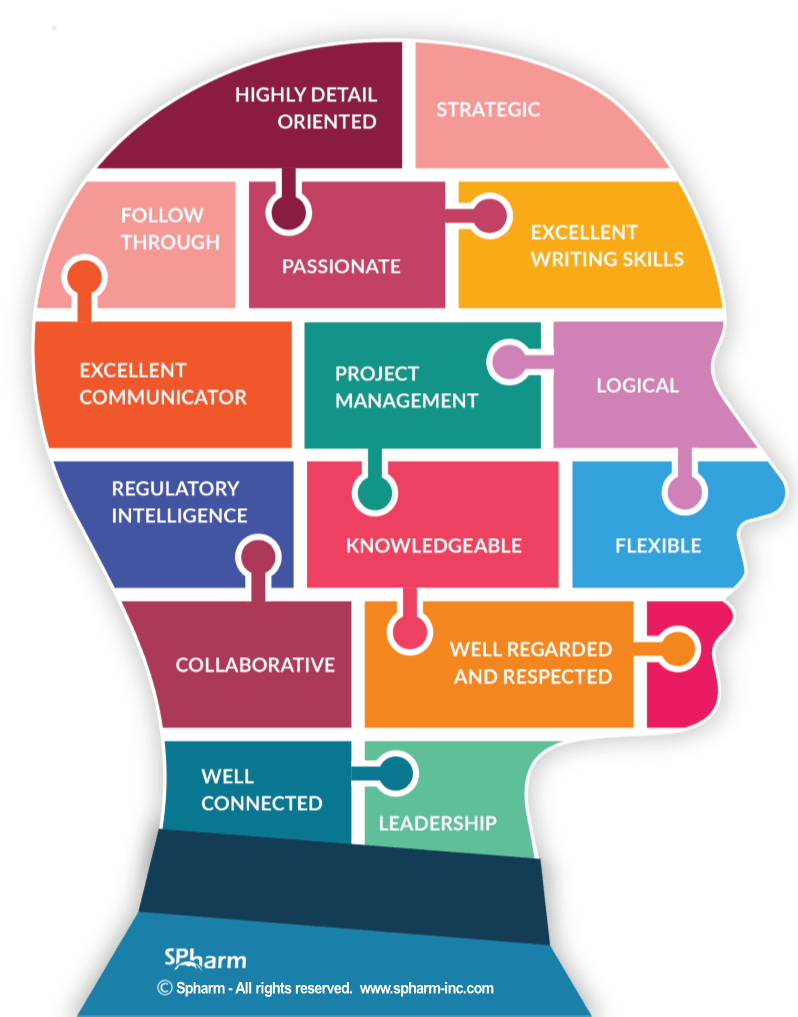
We have often been tasked to “find solutions” to the problems that arise when a regulatory professional isn’t brought in early-on in the health product development process. As such, we thought we’d share with you what the role of a Regulatory Affairs consultant is and what traits and attributes you will want to see in the person or consultant you hire.
Hire a Regulatory Consultant Early
Studies that are not well-planned are at risk of being unacceptable from a regulatory authority perspective, and yet still get conducted. As the study proceeds, development is consequently slowed or expanded as people address requirements ad-hoc. Submissions with inappropriate or insufficient data are prepared, causing submission review delays and its associated costs. There may even be instances where there is a need to go back to the drawing board and start from scratch or close to.
If the rationale behind not bringing a regulatory professional on board early was savings, companies often discover too late that it may end up costing them more on the back end. So, the selecting of an excellent regulatory professional is key and bringing them on at the planning stage, is tantamount.
One question worth asking is: Is there a specific certification that makes someone an effective regulatory professional or is it experience that makes you an expert? Well, the RAC certification from RAPS (of which I am a proud co-author) is well reputed for expertise and is an important foundation. That being said, the patina that develops over time managing a variety of challenging regulatory dossiers makes the most effective experts, in my view.
GOOD, JUST ISN’T GOOD ENOUGH
Here, we look in more detail at the benefits an excellent regulatory professional brings to each stage of the regulatory process (because good just isn’t good enough when we’re talking about the time, money, and value that this industry requires).
STAGE 1: RESEARCH & DISCOVERY
An excellent regulatory professional ensures that the planned development of a health product adheres to international Regulatory Agency requirements with a clear market aim in mind; this includes planning that considers the possibility of expanding the development geographically and facilitating the process of doing – lending flexibility to a development that may need to expand or could benefit from expanding.
STAGE 2: PRE-CLINICAL RESEARCH
In this stage, a regulatory professional ensures that the pre-clinical and the quality (CMC) development programs meet both local and international requirements prior to a first in-man study. This is where the traits of the professional become particularly important. Thorough and detail oriented, a keen logistical ability and regulatory intelligence separate those who are good from those who are great.
STAGE 3: CLINICAL TRIALS AUTHORIZATION
With the aim of obtaining authorization to conduct clinical trials, at this stage the role of a regulatory professional is to ensure that both local and international requirements are met. A regulatory professional coordinates pre-submission meeting, performs efficient and strategic medical and regulatory writing, performs QA and reviews submission documents for local and global compliance, prepares submissions to Health Authorities and ensures regulatory interfacing with them during the preparation, review processes and afterwards as needed.
Considering the significant sums of money involved, it’s not enough to have someone who has done it before or knows what to do, since the attributes of the person/the team can affect outcomes.
STAGE 4: REGULATORY PROCESS AND APPROVAL FOR MARKET ACCESS
In this fourth stage, the regulatory professional provides support and knowledge during each step of the registration process. Specifically, your expert should assist you in successfully navigating submission preparation, identify potential concerns ahead of submission time and proactively find solutions. In this phase, expert should perform efficient and strategic medical and regulatory writing, QA and reviews submission documents for completeness as well as local and global compliance, interface with health authorities and makes the regulatory process more predictable until drug approval. Here also, expertise is key.
STAGE 5: COMPLEMENTARY INITIATIVES
Regulatory professionals act not only as guides through the drug development processes at the pre-clinical, clinical phases and registration for market access phases. Experts also prepare provincial reimbursement submissions and provide support in reimbursement negotiations with decision makers. They also review marketing material and advertisement initiatives for compliance with the Food and Drug Regulations and PAAB Code and liaise between PAAB decision makers and sponsor, activities that are also key in the market access profession (combination of provincial initiatives in addition to the Federal ones).
STAGE 6: ONGOING POINT OF CONTACT
When the registration process is complete, and a health product can reach the market, a regulatory professional remains the point of contact with Health Authorities, to ensure compliance behind maintaining a registration. A true regulatory expert will evaluate the reportability of pharmacovigilance activities in compliance with regulations, evaluate the type of post-approval variations and documents required, prepares and coordinates all post approval submissions to Health Authorities, and coordinates telecoms or face-to-face meetings with them as needed.
IN SUMMARY
Regulatory affairs professionals are your quintessential regulatory process concierge; experts to call on the moment you first begin thinking about planning any medical device, drug or other health product development so that each stage of the process is navigated smartly and skillfully.
Considering the significant sums of money involved in the development process of a drug, medical device or other health product, it is simply not worthwhile to hire or work with someone who is not considered an excellent regulatory expert.
With a quarter of a century of consistent and successful results, SPharm has become one of the leading Drug Regulatory Advisory firms in Canada. Drug companies from around the world count on SPharm to help them navigate the drug (and other health products) review & approval process in Canada.
Avec un quart de siècle de résultats et de succès constants , SPharm est devenue une entreprise de services de référence au Canada en stratégie de développement de médicaments et affaires réglementaires. Des compagnies pharmaceutiques à travers le monde comptent sur SPharm pour faciliter le processus d’homologation de leurs médicaments (et autres produits de santé) au Canada.












