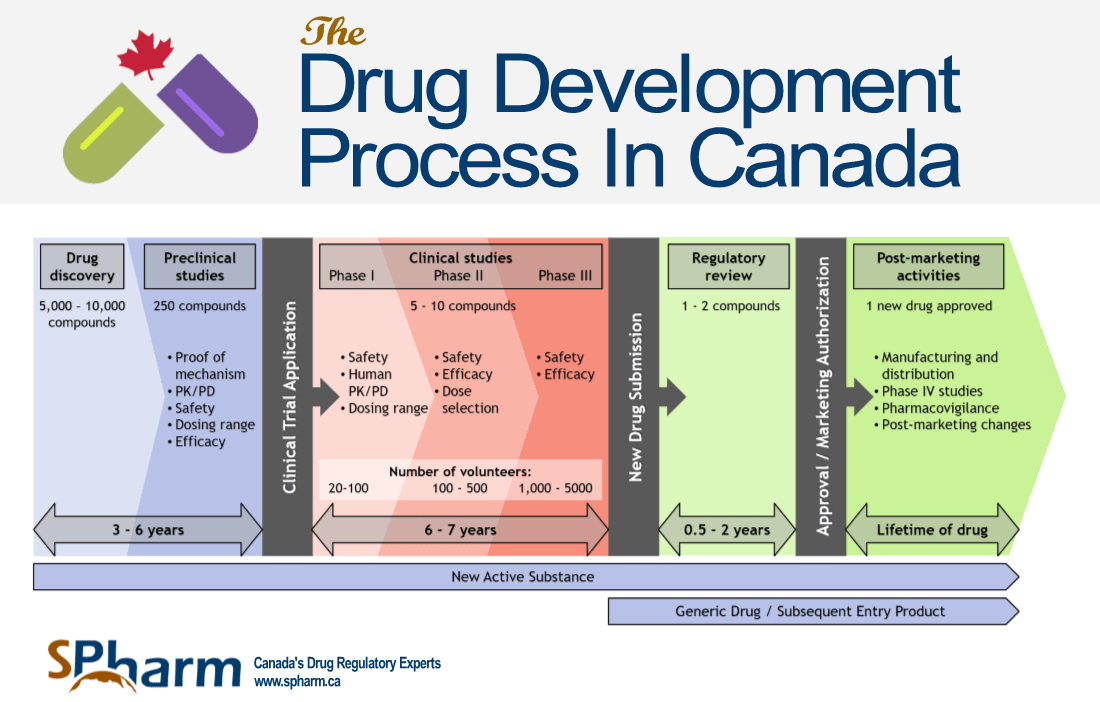
In Canada, as in all countries with regulatory agencies, there are specific regulatory pathways and processes for health products to be authorized for use in clinical trials or to obtain marketing approval. If you are looking to conduct a clinical trial or submit for the marketing authorization of a new health product, it is important to be familiar with the new drug submission process in Canada and what other regulatory processes look like, including the pathways that are available to you to achieve this.
Drug Regulatory affairs
Regulatory affairs in relation to clinical trials or investigational testing refers to the process of ensuring that a trial conducted with patients or healthy individuals adheres to all relevant laws and regulations during the drug, medical device, or other health product clinical development process. This includes getting necessary authorizations from regulatory agencies governing the country where the sponsors wish to launch a trial.
In Canada, that regulatory agency is Health Canada (HC). The goal of HC, its Food and Drug Regulations, and regulatory processes is to protect the health and safety of the participants in those trials and ensure the accuracy and integrity of the data collected; this includes the reporting of any serious adverse events that occur during the studies. HC is interested not only in the safety of trial participants; data accuracy and integrity are as important and are needed for the eventual market access application you will submit at the end of the clinical development process.
Most countries have their own regulating body, for example, the Food and Drug Administration (FDA) in the United States (US), European Medicines Agency (EMA) in the European Union, Therapeutic Goods Administration (TGA) in Australia and the Central Drugs Standard Control Organization (CDSCO) in India, to name a few.
What they have in common is that the approach to trials, drug development, and market access process be transparent and efficient whilst ensuring the safety, efficacy, and quality of the product for their citizens.
Regulatory and licensing pathways
In Canada, Health Canada has various licensing pathways for drugs, medical devices and other health products and they continue to work towards different pathways to facilitate market access of key health products, particularly those for unmet medical needs.
The regulatory strategies and submission work are done by regulatory experts; professionals well-versed and experienced in how each regulatory body evaluates and approves New Drug Submissions (NDS), Medical Devices License Applications as well as other health product applications in Canada or their equivalent outside Canada.
Specifically, the drug approval pathways in Canada are summarized as follows:
Regular New Drug Submission Process:
New Drug Submission is the regulatory process required by Health Canada that pharmaceutical companies are required to go through to bring their new drugs to the market. The submission must include all relevant data and information about the drug, including non-clinical, clinical, and quality data, which address safety and efficacy as well as the overall quality (chemistry, manufacturing, and controls) of that drug. Health Canada reviews this information to determine if the drug is of good quality, safe and effective (positive benefit vs. risk ratio) for its intended use. Only then can it be approved for sale to the public.
The standard timeframe for scientific review of the NDS is 300 days, preceded by a screening period of 45 days and 10 days of technical processing, as indicated below:

Priority Review New Drug Submission Process
A Priority Review (PR) New Drug Submission (NDS) is a type of regulatory filing submitted to Health Canada for the review of a new drug, at the end of its clinical development program, that is considered to have the potential to provide significant benefit over existing therapies (when available) for serious or life-threatening conditions. PR NDSs are prioritized over standard NDSs and are scientifically reviewed within 180 days, rather than the standard review time of 300 days. This expedited review process is intended to help bring new, innovative treatments to patients in need more quickly.
To apply for a Priority Review NDS, a sponsor must demonstrate that the drug is to be used for a serious, life-threatening, or severely debilitating condition when there is substantial evidence of clinical effectiveness that shows the following:
- effective treatment, prevention or diagnosis of a disease, for which no therapy is available in Canada, or
- significant increase of efficacy and/or decrease in risk, supporting an improved benefit/risk profile over available treatment, prevention, or diagnostic agent for a disease, not adequately managed by available agents in Canada.
In summary, Priority Review New Drug Submission Process in Canada allows for drugs with a complete clinical program that address important unmet medical needs. The sponsor has to apply for a PR through an official request supported by a Clinical Assessment Package, justifying scientifically and with sufficient data why the submission should be granted PR rather than following the standard NDS process.
The Priority Review process differs from the regular NDS process as presented below:

Notice of Compliance with Conditions – NOC/c NDS:
The Notice of Compliance with Conditions (NOC/c) pathway allows a sponsor to bring a new drug to market during the clinical development process, and therefore more quickly, in exchange for accepting to complete the clinical development program agreed to with Health Canada. An approval under the NOC/c policy contains conditions that a sponsor needs to comply with after the drug approval.
Eligibility
Eligibility for advanced consideration for a NOC/c applies to NDSs and SNDSs for serious, life-threatening, or severely debilitating conditions when there is promising evidence of clinical effectiveness based on the available data that shows the following:
- effective treatment, prevention, or diagnosis of a disease, for which no therapy is available in Canada, or
- significant increase of efficacy and/or decrease in risk, supporting an improved benefit/risk profile over available treatment, prevention, or diagnostic agent for a disease, not adequately managed by available agents in Canada.
NOC/cs are possible for drugs with promising clinical data that addresses important unmet medical needs. The NOC/c process differs from the regular NDS process as presented below:

Drug Submissions Relying on Third-Party Data (Literature and Market Experience):
This refers to the use of existing scientific literature or third party published data in lieu of clinical study reports to support the safety and efficacy of a drug. Literature can be used to support a New Drug Submission (NDS) or a Supplemental New Drug Submissions (SNDS) in Canada.
This approach saves time and resources for both the sponsor and HC, provided that the submission complies with the criteria established by HC. The literature must be relevant, reliable, and adequate to support the safety and efficacy of the drug in question and meet current regulations, guidelines and recommendations. The chemistry, manufacturing and controls (quality) data requirements are the same as for any other NDSs or SNDSs.
In addition to the pathways summarized above, HC does explore additional pathways to facilitate drug development to address unmet medical needs and to streamline their own processes, for example: Australia-Canada-Singapore-Switzerland (ACSS) Consortium and use of foreign reviews and decisions.
Project Orbis is another example of an international partnership HC is a part of which is designed to give cancer patients faster access to promising cancer treatments, alongside the US FDA. Products eligible for Project Orbis include oncology products that are either new active substances or new indications for previously approved drugs. Project Orbis submissions are expected to meet the criteria for “FDA Priority Review” and Health Canada PR or NOC/c criteria.
The various programs or pathways presented above can be explored with the assistance of regulatory experts, when a sponsor wishes to obtain a marketing authorization for a new drug in Canada. The various submission types have to respect specific criteria established by HC, which further underscores the need to work with regulatory experts that can orient the sponsor towards the most relevant and efficient pathway to use.
After the Notice of Compliance – NOC
Once Health has reviewed the submission and determined that the drug is safe and effective for its population, they will issue a Notice of Compliance (NOC), which grants the sponsor permission to sell the drug in Canada.
After an NOC has been granted for a new drug in Canada, maintaining the drug on the market requires various compliance activities, including pharmacovigilance and lifecycle management to ensure that the drug continues to be compliant with the requirements of the Food and Drug Regulations. Life cycle management activities could include a Supplemental New Drug Submission with new data from clinical trials to support a new indication or other important changes to the labelling of the drug. Likewise, major changes to the manufacturing process, or adding new formulations or a new dosage form, for example, would also require a submission to HC to ensure compliance with the regulations. For major changes that require approval from HC, a new NOC will be issued once HC has reviewed the submission and supporting data and, determined that the changes are acceptable; only then these changes can be implemented.
The Role of Regulatory Experts
Whichever pathway you utilize, it is imperative that the approach be one that is streamlined, transparent, and efficient, while ensuring the quality, safety, and efficacy of the product. It is the role of regulatory experts to strategize with you about which pathway to follow, and to navigate these applications through HC or their equivalent outside of Canada, to liaise with regulatory body officials, and lead the submission process.
For further information about the drug review process, please contact SPharm directly.



With a quarter of a century of consistent and successful results, SPharm has become one of the leading Drug Regulatory Advisory firms in Canada. Drug companies from around the world count on SPharm to help them navigate the drug (and other health products) review & approval process in Canada.
Avec un quart de siècle de résultats et de succès constants , SPharm est devenue une entreprise de services de référence au Canada en stratégie de développement de médicaments et affaires réglementaires. Des compagnies pharmaceutiques à travers le monde comptent sur SPharm pour faciliter le processus d’homologation de leurs médicaments (et autres produits de santé) au Canada.