GLOSSAIRE RÉGLEMENTAIRE DES MÉDICAMENTS AU CANADA
Pourquoi tous ces acronymes et définitions?
L’industrie pharmaceutique est complexe. Les termes, expressions, abréviations et le jargon propre à l’industrie sont également complexes et déroutants. Nous avons tenté de mettre de l’ordre dans tout ceci en préparant un glossaire des abréviations et des termes les plus couramment utilisés dans l’industrie.
Tous les acronymes ont été classés par ordre alphabétique.
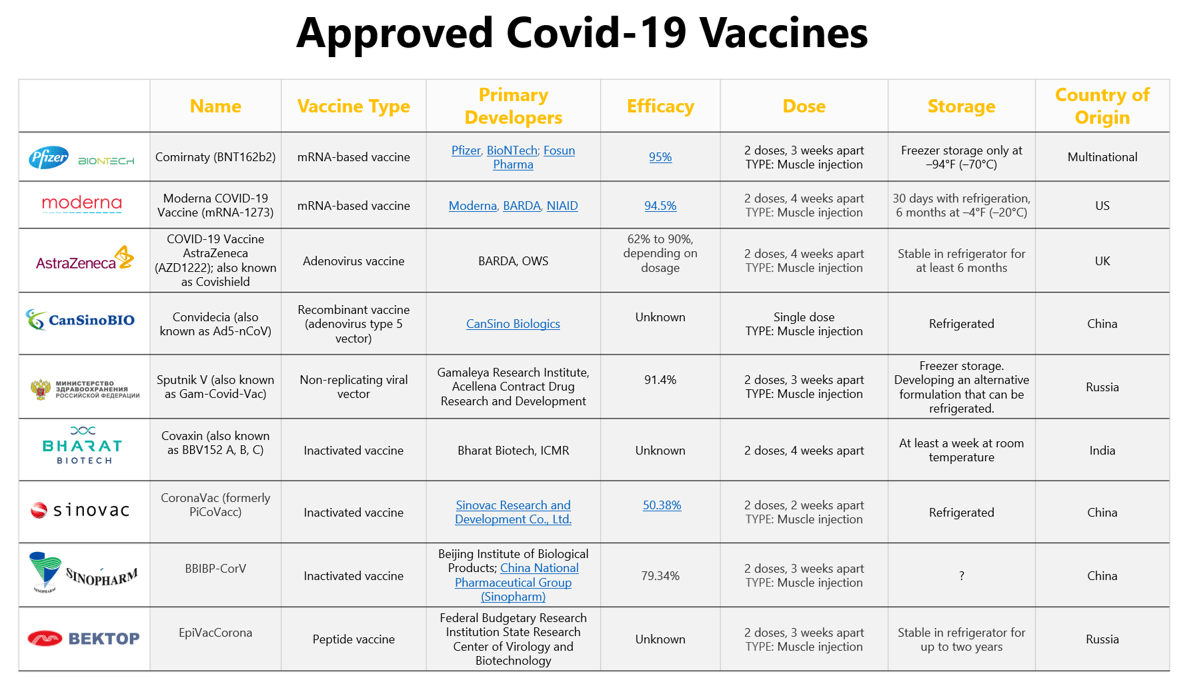
DAM – Déclaration annuelle des médicaments
Déclaration envoyée à Santé Canada chaque année, avant octobre, par un fabricant ou un promoteur de médicament, confirmant que tous les renseignements fournis antérieurement à l’égard de ce médicament sont exacts, conformément à l’article C.01.014.5 du Règlement sur les aliments et drogues. Le formulaire de déclaration annuelle des médicaments (FDAM) est disponible pour aider les fabricants à se conformer auxdites réglementations.
RID – Réaction indésirable à une drogue
Réaction nocive et non intentionnelle à une drogue qui survient lorsque la drogue est utilisée selon les doses normales ou selon des doses expérimentales, aux fins du diagnostic, du traitement ou de la prévention d’une maladie ou de la modification d’une fonction organique. Cela inclut l’absence d’effet.
IT – Incident thérapeutique
Événement indésirable affectant la santé d’un sujet d’essai clinique à qui une drogue a été administrée — qui peut ou non être causé par l’administration de la drogue, — y compris toute réaction indésirable à une drogue.
PADN – Présentation abrégée de drogue nouvelle
Dossier réglementaire devant être soumis et approuvé par Santé Canada pour qu’un fabricant obtienne l’autorisation de vendre (autorisation de mise en marché) un médicament générique sur le marché canadien. Ce dossier nécessite généralement une étude de bioéquivalence ou une comparaison physico-chimique (ou les deux) avec le produit de référence canadien (innovateur), ainsi qu’un dossier complet de chimie et de fabrication (qualité). Certaines exceptions peuvent s’appliquer.
EI – Effet Indésirable
Réaction nocive et non intentionnelle à un produit de santé commercialisé, incluant les expressions « réaction indésirable à une drogue », selon la définition du Règlement sur les aliments et drogues, et « incident thérapeutique », d’après le Règlement sur les produits de santé naturels. Dans ces dernières, un IT signifie une réaction nocive et non intentionnelle au produit de santé naturel, selon les doses normales ou selon des doses expérimentales, aux fins du diagnostic, du traitement ou de la prévention d’une maladie ou de la modification d’une fonction organique.
DMBR – Direction des médicaments biologiques et radiopharmaceutiques
Autorité réglementaire canadienne qui réglemente les médicaments biologiques (produits de sources vivantes) et les produits radiopharmaceutiques (contenant des éléments radioactifs) destinés à un usage humain au Canada. C’est l’une des directions de la Direction générale des produits de santé et des aliments. Avant qu’un fabricant ou un promoteur puisse commercialiser un produit, la direction doit prendre connaissance des preuves scientifiques de son innocuité, de son efficacité et de sa qualité, tel que l’exigent la Loi et le Règlement sur les aliments et drogues du Canada.
ACMTS – Agence canadienne des médicaments et des technologies de la santé
Organisme indépendant sans but lucratif dont le mandat est de fournir aux décideurs du système de santé des preuves objectives leur permettant de prendre des décisions éclairées concernant l’usage optimal des technologies de santé, notamment : les médicaments sur ordonnance, les tests diagnostiques, les procédures et dispositifs médicaux, dentaires et chirurgicaux. En plus des preuves, l’agence fournit également des conseils, des recommandations et des outils.
Créée en 1989 sous le nom de l’Office canadien de coordination de l’évaluation des technologies de la santé (OCCETS), l’ACMTS a adopté son nouveau nom en avril 2006 afin de mieux refléter ses activités globales.
DÉC – Dossier d’évaluation clinique
Document à préparer et à soumettre au bureau d’examen concerné de Santé Canada, accompagné d’une demande écrite d’obtention du statut d’examen prioritaire, avant le dépôt de la présentation de drogue nouvelle (PDN).
Produit de catégorie IV
Les produits de catégorie IV font référence à des produits dotés de monographies de catégorie IV, développés pour des médicaments dont le profil d’innocuité et d’efficacité est bien caractérisé dans des conditions d’utilisation spécifiques. Un fabricant peut faire référence à une monographie de catégorie IV dans une présentation de drogue lorsque le produit et son étiquetage sont conformes aux informations contenues dans le document. Les produits soumis aux monographies de catégorie IV peuvent obtenir un DIN par le biais d’une DDINF.
SCS – Société canadienne du sang
La Société canadienne du sang a été fondée en 1998 à la suite des recommandations du rapport Krever sur le scandale du sang contaminé du début des années 1990. La Société canadienne du sang est réglementée par Santé Canada en tant que fabricant de produits biologiques et financée principalement par les ministères de la Santé des provinces et des territoires. La Société canadienne du sang est un organisme à but non lucratif.
ICIS – Institut canadien d’information sur la santé
Organisme de santé canadien qui recueille et diffuse des données cliniques et non cliniques.
CIOMS – Conseil des organisations internationales des sciences médicales
Le CIOMS fait essentiellement référence au formulaire CIOMS I, qui fournit un format normalisé pour la déclaration des incidents thérapeutiques soupçonnés à tout produit médical, au Canada et dans le monde entier. Le Conseil joue un rôle important dans la pratique actuelle de pharmacovigilance.
Programme commun d’évaluation des médicaments (PCEM)
Processus national d’examen des médicaments non oncologiques qui met l’accent sur le rapport coût-efficacité d’un médicament par rapport aux traitements actuels. Le PCEM adresse une recommandation aux régimes publics d’assurance-médicaments (sauf au Québec, voir INESSS) à savoir si un médicament devrait ou non être financé par des fonds publics pour les patients qui en ont besoin.
EC – Essai clinique
Recherche sur des sujets humains dont l’objectif est de découvrir ou de vérifier les effets cliniques, pharmacologiques ou pharmacodynamiques d’un médicament, de déceler les incidents thérapeutiques liés à ce médicament, d’en étudier l’absorption, la distribution, le métabolisme et l’élimination ou d’en établir l’innocuité ou l’efficacité.
DEC – Demande d’essai clinique
Dossier réglementaire devant être soumis à Santé Canada et afin d’obtenir une lettre de non-objection (LNO) de Santé Canada avant que le promoteur puisse démarrer un essai clinique sur un produit pharmaceutique, biologique ou radiopharmaceutique à l’étude chez la population ou des patients canadiens.
En anglais, DEC devient CTA, abréviation qui peut avoir plusieurs significations. Dans le contexte d’activités de gestion d’une étude clinique, CTA peut également vouloir dire “Clinical Trial Agreement”, c’est-à-dire une entente entre le site de l’étude et le promoteur afin que ladite étude soit menée.
CTA peut également signifier “Clinical Trial Authorization”, ce qui correspond à l’équivalent européen (EMA) d’une DEC. Aux États-Unis (FDA), un IND (« Investigational New Drug ») est l’équivalent d’une demande d’essai clinique au Canada.
MDEC et NDEC – Modification et notification de demande d’essai clinique
MDEC : Changements apportés au protocole ou au dossier qualité, après la soumission de la DEC originale qui modifient la sécurité des patients, l’analyse des résultats et l’interprétation de l’efficacité et l’innocuité du médicament à l’étude, ou qui pourraient compromettre la qualité et la sécurité des approvisionnements du médicament à l’étude. Une période d’examen de 30 jours s’applique avant que ces changements ne puissent être mis en œuvre.
NDEC : Changements apportés ne correspondant pas aux critères d’une MDEC et qui peuvent être mis en œuvre immédiatement. Le promoteur doit cependant en aviser Santé Canada par écrit dans les 15 jours qui suivent la date de mise en œuvre des changements.
CTD – Common Technical Document
Format de demande standardisé mondialement et accepté dans plusieurs régions, dans le but d’éviter de devoir compiler différents dossiers d’homologation pour différentes autorités réglementaires. Le format CTD a été adopté par Santé Canada en 2003.
LEC – Lieu de l’essai clinique
Lieu où les activités liées à l’essai clinique sont effectivement menées.
FILEC – Formulaire d’information sur le lieu d’essai clinique
Formulaire que le promoteur ou son représentant doit remplir et soumettre à Santé Canada pour chacun des sites d’essai clinique avant le début de l’essai ou de la mise en œuvre d’une modification à la demande d’essai clinique à l’un de ces sites.
LEPP – Licence d’établissement de produits pharmaceutiques
Licence émise à une personne au Canada l’autorisant à mener des activités requérant une licence dans un bâtiment inspecté et jugé conforme aux exigences du Règlement sur les aliments et drogues. Les activités couvertes en vertu d’une LEPP sont la fabrication, l’emballage, l’étiquetage, les tests, l’importation, la distribution ou la vente en gros. Une licence d’établissement de produits pharmaceutiques s’applique pour les principes actifs de médicaments, les médicaments sous leur forme galénique finie ou les produits intermédiaires en vrac.
DIN – Numéro d’identification d’un médicament
Numéro unique à 8 chiffres assigné à chaque produit médicamenteux approuvé en vertu de la Loi et du Règlement sur les aliments et drogues (à l’exception des produits radiopharmaceutiques d’annexe C). Un DIN permet d’associer un produit aux caractéristiques suivantes: fabricant, marque nominative, ingrédient(s) médicinal(aux), dosage, forme galénique et voie d’administration.
DDIN – Demande d’identification numérique de drogue
Lorsqu’un produit n’est pas sujet au Titre 8 du Règlement sur les aliments et drogues, la requête est appelée demande d’identification numérique de drogue (DIN).
Note : Sous les dispositions de la section C.01.014 du Règlement sur les aliments et drogues, aucun fabricant ne doit vendre un médicament sous une forme galénique sans que celui-ci se soit vu attribuer un DIN ou qu’un DIN lui ayant été attribué ait été annulé en vertu de la section C.01.014.6. Dans le cas d’un nouveau médicament, la soumission d’une présentation de drogue nouvelle est considérée, en vertu du Titre 8 du Règlement sur les aliments et drogues, comme une demande de DIN.
Un DDINB est une demande de DIN spécifique aux produits biologiques.
Un DDIND est une demande de DIN spécifique aux produits désinfectants.
Un DDINF est une demande de DIN spécifique aux produits de catégorie IV.
LAD – Loi sur les aliments et drogue
Loi encadrant les aliments, les médicaments, les cosmétiques et les instruments thérapeutiques au Canada.
RAD – Règlement sur les aliments et drogues
Règlement qui encadre et établit les exigences pour la fabrication, l’emballage, l’étiquetage, l’entreposage, l’importation, la distribution et la vente d’aliments et de médicaments sur ordonnance ou en vente libre au Canada. Les exigences concernant les études cliniques sur des médicaments sont également encadrées par ce règlement. Santé Canada développe et met en application cette réglementation sous l’autorité législative du gouvernement du Canada. Le ministère consulte le public canadien, l’industrie et d’autres parties d’intérêt concernant l’élaboration de lois visant à protéger la santé et la sécurité. Il rédige et publie également des lignes directrices et des politiques afin de faciliter l’interprétation et ainsi clarifier toute la législation entourant les médicaments et tout autre produit de santé. Ce règlement a pour but de protéger la santé et la sécurité des Canadiens en ce qui concerne la vente d’aliments et de médicaments.
BPC – Bonnes pratiques cliniques
Pratiques cliniques standardisées généralement acceptées conçues pour assurer la protection des droits, de la sécurité et du bien-être des sujets d’essais cliniques ou de toute autre personne concernée. Les bonnes pratiques cliniques s’appliquent en vertu de la section C.05.010 du Règlement.
BPF – Bonnes pratiques de fabrication
Aspect du système qualité qui s’assure de la régulation et du contrôle de la production des médicaments de façon à ce qu’ils respectent les normes de qualité appropriées selon leur usage prévu, tel qu’exigé par l’autorisation de mise en marché. Une partie du programme de l’Inspectorat de la Direction générale des produits de santé et des aliments consiste à effectuer l’inspection des établissements exerçant des activités comprises dans le cadre d’une licence d’établissement.
SC – Santé Canada
Ministère fédéral chargé d’aider les Canadiens à maintenir et améliorer leur santé tout en respectant les choix individuels et les circonstances.
PS – Produit de santé
Produit régulé en vertu du Règlement sur les aliments et drogues (médicaments) et du Règlement sur les produits de santé naturels (produits de santé naturels). Les médicaments comprennent aussi bien les médicaments sur ordonnance qu’en vente libre; les produits biotechnologiques et d’origine biologique tels que les vaccins, les sérums et les produits dérivés du sang; les désinfectants; les produits radiopharmaceutiques.
DGPSA – Direction générale des produits de santé et des aliments
Division de Santé Canada ayant pour mandat de gérer les risques pour la santé et les bienfaits des produits de santé et des aliments par les moyens suivants : 1) minimiser les facteurs de risque pour la santé des Canadiens tout en maximisant l’innocuité qu’assure le système réglementaire encadrant les aliments et les produits de santé; 2) rendre l’information accessible aux Canadiens afin de leur permettre de faire des choix sains et éclairés. Les activités de la DGPSA s’exercent sous diverses directions, notamment la Direction des produits thérapeutiques, la Direction des médicaments biologiques et radiopharmaceutiques et la Direction des produits de santé commercialisés.
IDGPSA – Inspectorat de la Direction générale des produits de santé et des aliments
Une partie du programme de l’IDGPSA consiste à mener des inspections d’établissements exerçant des activités comprises dans le cadre d’une licence d’établissement. Ces inspections visent à vérifier la conformité avec les BPF (Partie C, Titre 2 du Règlement sur les aliments et drogues), ce qui est l’une des exigences pour l’obtention d’une licence d’établissement.
HQ – Héma Québec
Société à but non lucratif, Héma-Québec a été constituée en 1998, des longues suites du rapport final de la commission Krever, initié en raison du scandale du sang contaminé au début des années 90. En matière de sécurité de l’approvisionnement, Héma-Québec est une organisation nationale, pour le Québec, légiféré par Santé Canada comme fabricant de produits du sang (biologiques).
ETS – Évaluation des technologies de la santé
Processus à suivre pour formuler des recommandations fondées sur des faits à savoir si une technologie de la santé mérite ou non d’être financée à même les fonds publics.
BI – Brochure de l’investigateur
En ce qui concerne un médicament, il s’agit d’un document contenant les données cliniques et non cliniques au sujet du médicament, tel que décrit à la section C.05.005(e) du Règlement.
FCE – Formulaire de consentement éclairé
Document décrivant à un sujet recruté pour un essai clinique les risques et bénéfices anticipés pour sa santé découlant de sa participation à l’essai clinique, ainsi que tout autre aspect de l’essai nécessaire pour lui permettre de prendre une décision éclairée quant à son choix de prendre part ou non à l’essai.
ICH – International Conference on Harmonization
Initiative internationale visant à harmoniser mondialement les exigences relatives à l’efficacité, l’innocuité et la qualité (chimie et fabrication) pour l’homologation des médicaments (pharmaceutiques, biologiques, radiopharmaceutiques) destinés à l’usage chez l’humain. Cette initiative comprend une organisation standardisée de l’information pour les demandes d’homologation de drogues nouvelles.
INESSS – Institut national d’excellence en santé et en services sociaux
Organisme québécois assurant les processus d’examen afin d’évaluer la valeur thérapeutique et le rapport coût-efficacité des médicaments oncologiques et non oncologiques. L’INESSS transmet des recommandations au ministère de la Santé et des Services sociaux du Québec à savoir si un médicament devrait être financé par les fonds publics pour les patients en ayant besoin.
DEE – Demande d’essai expérimental
L’équivalent d’une DEC pour les instruments médicaux.
TAMM – Titulaire de l’autorisation de mise en marché
L’entité légale détenant l’Avis de conformité, le numéro d’identification du médicament (DIN), le numéro de produit naturel (NPN), le numéro de produit homéopathique (DIN-HM), ou la licence d’un produit.
DHIM – Demande d’homologation d’un instrument médical
L’équivalent d’une DEC, mais pour étudier l’efficacité et l’innocuité d’un dispositif médical.
FM – Fiche maîtresse
Anciennement nommé « Fiche maîtresse d’un médicament ». Référence fournissant de l’information sur des procédés particuliers ou composants utilisés dans la fabrication, le traitement et l’emballage d’un médicament. La FM est un outil utile pour transmettre de l’information à Santé Canada, surtout lorsqu’il s’agit de renseignements exclusifs (c’est-à-dire des renseignements corporatifs confidentiels), et non accessibles au fabricant de la forme galénique ou au promoteur d’une présentation de drogue ou d’une DEC (aussi appelés les « demandeurs »).
NSA – Nouvelle substance active
- Substance chimique ou biologique dont la vente en tant que médicament au Canada n’avait pas été autorisée;
- Isomère, dérivé ou sel d’une substance chimique dont la vente en tant que médicament au Canada a déjà été autorisée, mais dont les propriétés varient au niveau de l’innocuité et de l’efficacité;
- Substance biologique dont la vente en tant que médicament au Canada a déjà été autorisée, mais dont la structure moléculaire, la nature de la matière première ou les procédés de fabrication diffèrent.
PM – Préavis de modification
Les préavis de modification sont des préavis de niveau 2 et classés soit comme : des modifications de qualités modérées (chimique et manufacturiers) qui ont un potentiel modéré d’avoir des effets indésirables sur l’identité, la concentration, la qualité, la pureté ou l’activité du médicament, car ces facteurs peuvent être liés à l’innocuité ou à l’efficacité du médicament. Ce niveau de préavis ne s’applique pas aux produits pharmaceutiques destinés aux humains
Une modification de gestion du risque (clinique) est définie comme un changement à l’étiquetage qui a le potentiel d’améliorer la gestion du risque dans la population cible ou autrement exposée au médicament. Ces changements sont classés soit comme des changements en examen de 90 jours (changements plus urgents) ou des changements d’examen de 120 jours.
PDN – Présentation de drogue nouvelle
Dossier réglementaire devant être soumis et approuvé par Santé Canada pour qu’un fabricant obtienne l’autorisation de vendre (autorisation de mise en marché) un médicament générique sur le marché canadien (c.-à-d. pharmaceutique, biologique, vaccin, produits biotechnologiques). Ce dossier nécessite généralement un dossier pré-clinique, clinique et chimie et fabrication (qualité) complet. Des exigences spécifiques peuvent différer selon la catégorie de drogue, la pathologie traitée et la population de patients ciblée entre autres éléments.
PSN – Produits de santé naturels
Une substance figurant à l’annexe 1 du Règlement sur les produits de santé naturels ou une combinaison de substances dont tous les ingrédients médicinaux sont des substances figurant à l’annexe 1 du Règlement sur les produits de santé naturels, une médecine homéopathique ou un médicament traditionnel.
AV – Avis de conformité
Avis émis par Santé Canada indiquant qu’un fabricant s’est conformé aux exigences du Règlement sur les aliments et drogues à la fin d’un examen d’une PDN, PADN, SPDN et SPADN
AC-C/AA – Avis de conformité avec conditions – Avis d’admissibilité
Avis émis par le bureau/centre responsable à la fin d’un examen, lorsqu’une présentation est jugée admissible à un examen ultérieur en vertu de la politique AC-C (NOC/c). L’AC-C/AA indiquera que la présentation est admissible à un AC, en vertu de la politique AC-C, et décrira les preuves cliniques supplémentaires à fournir dans les études de confirmation, les responsabilités de surveillance post-commercialisation et toute exigence liée à la publicité, l’étiquetage ou la distribution. L’examen de la présentation cessera dès la publication de l’avis d’admissibilité.
Cela s’applique aux produits qui présentent des données prometteuses d’efficacité clinique ainsi qu’un profil d’innocuité acceptable destinés au traitement, à la prévention ou au diagnostic d’une maladie ou d’une affection grave, potentiellement mortelle ou très invalidante pour laquelle il n’existe aucun traitement au Canada, ou pour laquelle la présentation de drogue nouvelle démontre une amélioration significative du profil bénéfices/risques par rapport au produit alternatif disponible.
AI – Avis d’insuffisance
Un avis d’insuffisance est émis si des lacunes et/ou des omissions importantes empêchent la poursuite de l’examen d’une présentation.
LNO – Lettre de non-objection
Lettre émise par Santé Canada après l’examen d’une demande d’essai clinique ou d’un préavis de modification, si la demande est jugée recevable. Elle confirme qu’un promoteur peut procéder à son essai clinique au Canada ou peut mettre en œuvre les modifications présentées dans le préavis de modification.
ANC – Avis de non-conformité
Avis émis après l’examen minutieux d’une présentation, si celle-ci est insuffisante ou incomplète et par conséquent non conforme aux exigences énoncées dans la Loi et le Règlement sur les aliments et les drogues.
ANS – Avis de non-satisfaction
Avis émis par le bureau/centre responsable si des lacunes sont décelées lors de l’examen d’une demande d’essai clinique, ou d’une modification ou préavis de modification à une demande d’essai clinique. Les lacunes seront précisées et l’examen de la présentation cessera à la date de l’avis de non-satisfaction.
CCPP – Conseil consultatif de publicité pharmaceutique
Le Conseil consultatif de publicité pharmaceutique (CCPP) est un organisme indépendant et à but non lucratif qui est financé selon la formule du « paiement à l’acte ». Il s’agit du seul organisme de réglementation dont Santé Canada reconnaît le service de préagrément du matériel publicitaire qui s’adresse directement aux professionnels de la santé. Le CCPP travaille à la protection des Canadiens en veillant à ce que les publicités sur les produits de soins de santé soient conformes aux normes réglementaires, scientifiques, thérapeutiques et éthiques décrites dans le Code d’agrément de la publicité. Tout matériel approuvé par le CCPP affiche le logo du CCPP.
RPEAR – Rapport périodique d’évaluation des avantages et des risques
Document de pharmacovigilance destiné à fournir une analyse minutieuse, concise et critique de l’information nouvelle ou émergente sur les risques du produit de santé et sur son avantage dans les indications approuvées, afin de permettre une évaluation du profil bénéfice-risque global du produit. Les directives actuelles de l’ICH garantissent que les RPPV (rapports périodiques de pharmacovigilance) pour les médicaments commercialisés ont un rôle de rapports périodiques d’évaluation des avantages et des risques en couvrant : l’évaluation de l’innocuité, l’évaluation de tous les renseignements disponibles pertinents accessibles aux promoteurs/titulaires de l’AMM et l’évaluation des bénéfices-risques.
APP – Alliance pancanadienne pharmaceutique
Mécanisme national conçu pour accroître la valeur des régimes d’assurance-médicaments gouvernementaux. L’APP négocie avec une société pharmaceutique pour déterminer à la fois le coût et les critères selon lesquels les gouvernements paieront pour un médicament, concluant avec une lettre d’intention pour financer le médicament.
CEPMB – Conseil d’examen du prix des médicaments brevetés
Le CEPMB protège et informe les Canadiens en s’assurant que les médicaments brevetés ne sont pas vendus au Canada à des prix excessifs et en faisant rapport des tendances pharmaceutiques.
EP – Examen prioritaire
Statut accordé par Santé Canada à une PDN ou à un SPDN pour une maladie grave, potentiellement mortelle ou très invalidante pour laquelle il existe des preuves substantielles de l’efficacité clinique que le médicament fournit : 1) un traitement, une prévention ou un diagnostic d’une maladie ou affection pour laquelle aucun médicament n’est actuellement commercialisé au Canada; ou 2) une augmentation significative de l’efficacité et/ou une diminution significative du risque telle que le profil global bénéfice/risque est amélioré par rapport aux thérapies, agents préventifs ou agents diagnostiques existants pour une maladie ou un état qui n’est pas pris en charge de manière adéquate par un médicament commercialisé au Canada.
MEIEP-DEC- Modèle d’évaluation de l’innocuité et de l’efficacité des protocoles – demande d’essai clinique
Résumé du protocole à préparer et à soumettre dans le cadre de la DEC. Le résumé devrait contenir : l’identification du protocole, le contexte et la justification, les objectifs de l’essai, la conception et la durée de l’étude, le nombre total de sites (y compris le nombre de sites canadiens), les investigateurs, la taille de l’échantillon, la population de patients, les critères d’inclusion et d’exclusion, la formulation du médicament, le schéma posologique, la période de sevrage, les visites de traitement et d’évaluation, les médicaments concomitants et de secours, la gestion des risques, les critères de retrait/d’arrêt de l’essai, les variables et l’analyse d’efficacité et d’innocuité et l’analyse statistique de l’essai clinique.
RPPV – Rapport périodique de pharmacovigilance
Mécanisme pour la synthèse des données, compilées à intervalles donnés, sur l’innocuité et pour les évaluations globales de l’innocuité. Il s’agit d’un outil dont se servent les promoteurs pour l’analyse systématique des données sur l’innocuité effectuée sur une base régulière. En plus de traiter des questions courantes en matière d’innocuité, le RPPV doit également comporter des mises à jour sur les questions nouvelles ou urgentes relativement à l’innocuité et sur la détection et l’évaluation principales des signaux abordées dans d’autres documents.
PSQ – Professionnel de la santé qualifié
Personne membre en règle d’une association professionnelle de médecins, d’infirmières, de pharmaciens ou d’autres praticiens de la santé et autorisée à fournir des soins de santé en vertu des lois de la juridiction dans laquelle la personne se trouve, et d’autres personnes retenues par le titulaire de l’autorisation de mise en maché ayant la formation et l’expertise thérapeutique appropriées en soins de santé.
IQ – Investigateur qualifié
Personne responsable envers le promoteur de la conduite de l’essai clinique sur le site de l’essai clinique, qui est autorisée à fournir des soins de santé en vertu des lois de la province où ce site d’essai clinique est situé.
EIQ – Engagement de l’investigateur qualifié
Engagement que doit remplir l’investigateur qualifié responsable de la conduite de l’essai clinique au site de l’essai clinique et retenu par le promoteur de l’essai clinique pour une période de 25 ans. Cet engagement ne doit pas être soumis à Santé Canada à moins qu’il ne soit demandé.
RGQ – Résumé global de la qualité
Modèle de résumé qui suit la portée et les grandes lignes de l’ensemble des données sur la qualité. (du module CTD 3.2). Des modèles de RGQ spécifiques existent pour les phases I, II et III des demandes d’essais cliniques; pour les études de biodisponibilité et pour les demandes de DIN. Le RGQ pour les nouvelles entités chimiques est présenté ci-dessous.
RGQ-EC – Résumé global de la qualité- Entités chimiques (DEC)
Modèle de résumé qui suit la portée et les grandes lignes de l’ensemble de données sur la qualité (du module CTD 3.2). Ce modèle peut être utilisé par les promoteurs pour résumer les renseignements sur la qualité des présentations de drogue nouvelle (PDN) et des présentations abrégées de drogue nouvelle (PADN) contenant des substances médicamenteuses et leurs produits correspondants d’origine synthétique ou semi-synthétique qui sont déposés auprès de Santé Canada conformément à la partie C, titre 8 du Règlement sur les aliments et drogues.
Cela exclurait les présentations de médicaments biotechnologiques/biologiques (annexe D) et radiopharmaceutiques (annexe C). Néanmoins, dans les faits, le RCG-EC référencé est utilisé pour résumer les informations sur la qualité pour les présentations de drogue nouvelle (PDN) de l’annexe D; les sections du modèle sont ensuite harmonisées avec les exigences des directives de l’annexe D de la PDN (EP). Un résumé de l’information sur la qualité (RIQ) est disponible pour les produits pharmaceutiques, à la demande de Santé Canada.
CER – Comité d’éthique de la recherche
Organisme non affilié avec le promoteur, ayant pour mandat d’approuver le lancement et de mener des examens périodiques de la recherche biomédicale impliquant des sujets humains afin de s’assurer de la protection de leurs droits, de leur sécurité et de leur bien-être. L’organisme doit compter au moins cinq membres, qui sont en majorité des citoyens canadiens ou des résidents permanents en vertu de la Loi sur l’immigration, et être composé à la fois d’hommes et de femmes. Des critères supplémentaires s’appliquent.
AMR – Activités de minimisation des risques
Les activités de minimisation des risques sont des interventions destinées à prévenir ou à réduire la survenue d’effets indésirables associés à l’exposition à un médicament, ou à réduire leur gravité ou leur impact sur le patient si des effets indésirables surviennent. Ces mesures peuvent inclure des mises en garde sur l’étiquette ou des activités de minimisation au-delà de la routine, telles que du matériel éducatif pour les pourvoyeurs de soins de santé.
PGR – Plan de gestion des risques
Document décrivant un ensemble d’activités et d’interventions de pharmacovigilance conçues pour identifier, caractériser, prévenir ou minimiser les risques liés aux produits médicamenteux, et l’évaluation de l’efficacité de ces interventions (adopté de la définition que donne l’Agence européenne des médicaments à un système de gestion des risques.)
SPADN – Supplément à une présentation abrégée de drogue nouvelle
Identique au SPDN présenté ci-dessous, mais pour les produits génériques.
SPDN – Supplément à une présentation de drogue nouvelle
Présentation requise pour les modifications de qualité ou d’innocuité et d’efficacité (cliniques) de niveau I qui ont un potentiel important d’avoir des effets indésirables sur l’identité, la concentration, la qualité, la pureté ou l’activité du médicament, car ces facteurs peuvent être liés à l’innocuité ou à l’efficacité du médicament, ou un changement à l’étiquette d’un médicament qui a le potentiel d’augmenter les niveaux d’exposition au médicament, soit en augmentant la population qui est exposée, soit en augmentant l’exposition individuelle.
PAS – Programme d’accès spécial
Programme qui permet l’accès à des médicaments non commercialisés pour les praticiens traitant des patients atteints d’affections graves ou potentiellement mortelles lorsque des thérapies conventionnelles ont échoué, ne sont pas adaptées ou ne sont pas disponible. Le PAS autorise un fabricant à vendre un médicament qui ne pourrait autrement être vendu ou distribué au Canada. L’autorisation obtenue dans le cadre du PAS permet à un fabricant de vendre un médicament qui n’a pas encore été approuvé pour la vente au Canada. Les médicaments auxquels le PAS pourrait donner accès comprennent des produits pharmaceutiques, biologiques et radiopharmaceutiques dont la vente n’est pas approuvée au Canada.
RIGM – Réaction indésirable grave à un médicament
Réaction indésirable à une drogue qui nécessite ou prolonge l’hospitalisation, entraîne une malformation congénitale ou une invalidité ou incapacité persistante ou importante, met la vie en danger ou entraîne la mort.
RIG – Réaction indésirable grave
Réaction nocive et non voulue à un produit de santé naturel provoquée par celui-ci, quelle qu’en soit la dose, et qui nécessite ou prolonge l’hospitalisation, entraîne une malformation congénitale, une invalidité ou une incapacité persistante ou importante, met la vie en danger ou entraîne la mort.
CS – Cadre supérieur
Personne détenant le pouvoir de décision politique et opérationnelle le plus élevé avec le promoteur, ou qui est un représentant à qui on a délégué ce pouvoir à l’égard d’un essai clinique. Le CS doit fournir une attestation concernant la demande/modification d’essai clinique au moment du dépôt de la DEC à Santé Canada.
RIGI – Réaction indésirable grave et imprévue
Réaction indésirable grave dont la nature, la gravité ou la fréquence n’est pas indiquée dans les mentions de risque figurant dans la brochure de l’investigateur ou sur l’étiquette du médicament.
DPT – Direction des produits thérapeutiques
Organisme canadien de réglementation des médicaments sur ordonnance et des dispositifs médicaux à usage humain. Avant d’autoriser la vente d’un produit, la direction doit voir les preuves scientifiques de l’innocuité, de l’efficacité et de la qualité du produit, comme l’exigent la Loi et le Règlement sur les aliments et drogues.
RAPB – Rapport annuel sur un produit biologique
Chaque année, les fabricants de drogues biologiques (annexe D) doivent produire un Rapport annuel sur un produit biologique, conformément à la Ligne directrice à l’intention des promoteurs : Programme d’autorisation de mise en circulation des lots de drogues visées à l’annexe D (produits biologiques). Le rapport contient des informations de production sur les lots de substances médicamenteuses et de produits médicamenteux, y compris les méthodes d’essai et les résultats, les raisons de tout rappel et les mesures correctives prises, ainsi que d’autres informations post-commercialisation pertinentes.